Fastjacc
July 2021
FastJacc
When it comes to molecular high-throughput-screening, you often need to compare a given molecule of interest against a database of (potentially billions) and look for similarities. Existing open-source libraries that do this are slow, clocking in the order of ~1-.1ms per comparison. This is slow, when you get to the order of billions.
The solution? Go minimal, and put it all on GPU.
Rather than hold complex “molecule” objects in memory, extract the parts of interest and put them in a raw binary format. Take a given database of molecules, convert it to this format. The similarity we care about can actually be expressed in terms of set operations over (fixed maximum size) binary sets, so write a vectorized way to do that, utilizing as few opterations as possible. In our case, this results in the only operations being simple integer arithmetic, and the CUDA population_count intrinsic.
After that, run a custom CUDA kernel over the dataset.
__global__ void fastJacc(unsigned int *allMols, unsigned int *queries,
float *sims, int row, int size)
{
// take steps over each fingerprint in allmols
// aka grid-stride loop
int index = blockIdx.x * blockDim.x + threadIdx.x;
int stride = blockDim.x * gridDim.x;
// the cardinality is stored as the first int, always.
int cardX = queries[65 * row];
for (int i = index * 65; i < size * 65; i += 65 * stride) {
int totalSize = 0;
// unroll the loop into 16 loops of 4. 15% speedup.
#pragma unroll 4
for (int j = 1; j < 65; j++)
{
unsigned int x = queries[(65 * row) + j];
unsigned int y = allMols[i + j];
totalSize += __popc(x & y);
}
int cardY = allMols[i];
// alternate eq for jaccard score: (int(x, y) / |x| + |y| - int(x, y)).
int jaccDenom = cardX + cardY - totalSize;
sims[(size * row) + (i / 65)] = float(totalSize) / jaccDenom;
}
}
Overall speedup? On the order of 300x, even including the cost of moving from CPU->GPU RAM and reading off disk.
The project also offers functionality to extract just the top-k most similar molecules in the database, in addition to supporting batching of inputs. Written entirely in C/C++/CUDA, getting better performance than this while still having exact solutions may only be possible in small amounts.
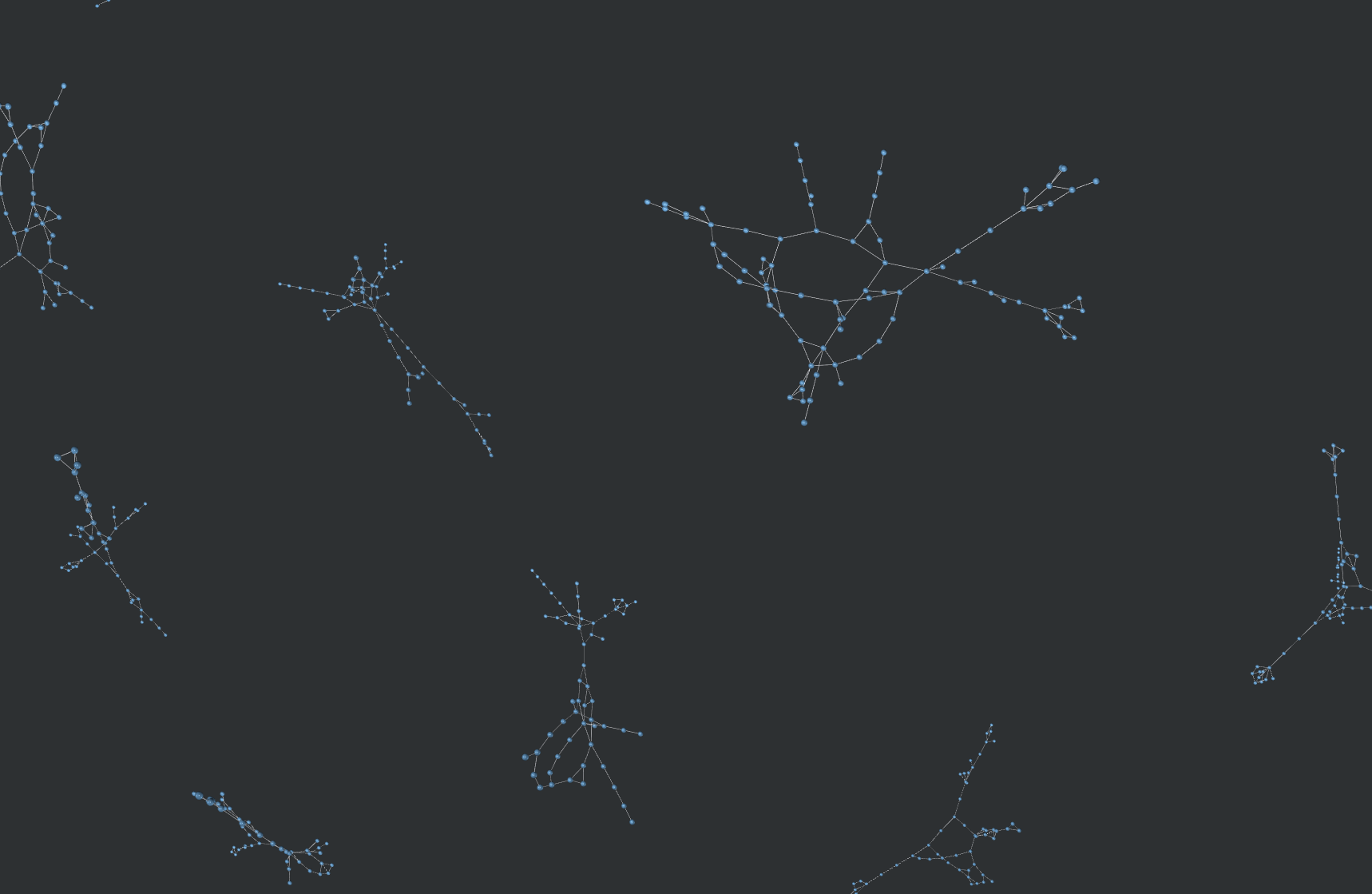
Below is an example of why this was needed. The following graphic compares the similarity of all 800k molecules within a dataset with eachother, resulting in 640,000,000,000 (640 billion) similarity computations. An edge exists between molecule A and molecule B if B is in the top-5 nearest neighbors of A.
With my implementation, this took a couple hours. On CPU, this would take a couple months.
Graph vizualized using Graphia and NetworkX.